Introduction
In this post, I would like to highlight a very recent paper titled: “You cannot fight the pressure: Structural rearrangements of active pharmaceutical ingredients under Magic Angle Spinning“. I find the article very interesting and important. Nuclear Magnetic Resonance (NMR) spectroscopy is widely assumed to be non-invasive and non-destructive. However, this paper shows that this might not be completely true in the case of solid-state NMR experiments performed on samples spinning under the Magic Angle. Fast Magic Angle Spinning (MAS) induces significant pressure in the sample. This may lead to phase transitions of pharmaceutical molecules and products.
Why should you care? If you study polymorphism of Active Pharmaceutical Ingredients (APIs) or stability of pharmaceutical formulations, the information here and in the original paper will be very useful to you. You need to be aware that MAS may alter your sample by inducing structural rearrangements and phase transitions.
Why solid-state NMR is used in pharmaceutical research and industry
Powder X-Ray Diffraction (powder XRD) is traditionally used to study polymorphism and control quality of APIs and pharmaceutical products. However, as powder XRD depends on the crystalline long-range order, it has very limited use in the case of amorphous phases. Since solid-state NMR does not require long-range order, it is an excellent tool for all solid samples, including crystalline and amorphous phases. For example, this JEOL application note demonstrates that solid-state NMR can differentiate between crystalline and amorphous forms of indometacin.
Why Magic Angle Spinning
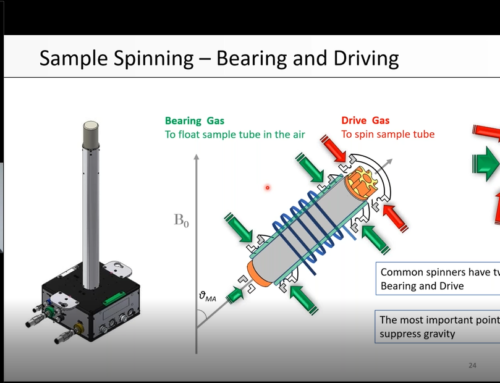
Magic Angle Spinning helps to increase resolution in solid-state NMR by mimicking the Brownian motion present in liquid samples. It suppresses anisotropic interactions such as dipolar interactions, Chemical Shift Anisotropy (CSA) and quadrupolar interactions. Please refer to our webinars for more details.
What Magic Angle Spinning does to solid samples
To give you a better appreciation of the spin rates and speeds used in solid-state NMR, let’s consider a 3.2 mm rotor spinning at 20 kHz. The circumference of the rotor is 3.2 × π ≈ 10 mm. If we multiply 10 mm by 20,000 revolutions, this gives a distance of 200,000 mm, or 200 m. This means that speed at the circumference is 200 m/s. For comparison, sound travels with the speed of 343 m/s in air. This is really fast!
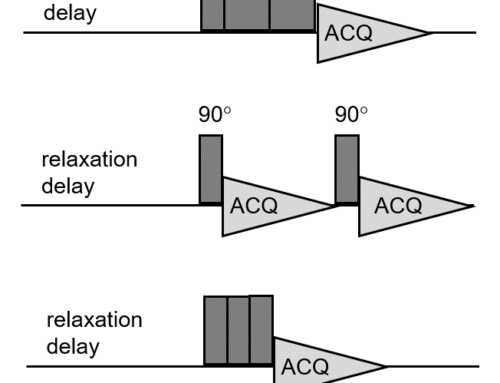
Somewhat unsurprisingly, fast MAS induces pressure and heating. Sample heating can be compensated for by blowing cold gas onto the spinning rotor as I described in a previous post. This also compensates for heating caused by the strong radiofrequency pulses irradiated during the solid-state NMR experiments. However, the only way to reduce the pressure is to spin slowly or not to spin at all. Unfortunately, this usually results in spectra of limited resolution.
It is important to note that MAS does not usually cause damage to typical solid samples. The majority of solid samples are resistant to the pressures induced by MAS. However, APIs are often favoured in amorphous forms for their improved solubility and bioavailability. Unfortunately, amorphous forms tend to be metastable, because they lack the long-range order. They may even spontaneously recrystalize into more stable crystalline forms upon storage. Stabilizing amorphous phases can be done by dispersing them into polymer matrices. Such pharmaceutical products are known as solid dispersions (SD) or, more specifically, amorphous solid dispersions (ASD). This application note demonstrates that ultrafast solid-state NMR can reveal drug-polymer interactions in solid dispersions.
What was shown in the paper
The authors studied atorvastatin calcium, ezetimibe and efivirenz. The former two treat hypercholesterolemia, whilst efavirenz treats HIV. The authors used powder XRD, ¹H MAS, ¹³C CPMAS, 19F MAS and ¹H-¹³C FSLG HETCOR (Frequency-Shifted Lee-Goldburg HETeronuclear CORrelation) to investigate the influence of MAS and storage on the samples.
They observed:
- Small changes in the ¹H-¹³C FSLG HETCOR spectrum of amorphous atorvastatin calcium collected after 12-24 hours of MAS. Powder XRD and 1D solid-state NMR spectra did not detect any obvious changes.
- Starting crystallization of amorphous ezetimibe after 12-24 hours of MAS by powder XRD and ¹H-¹³C FSLG HETCOR. The crystallization process progressed significantly during the ensuing storage without further sample spinning.
- Structural rearrangement in crystalline ezetimibe which was spun under the Magic Angle. Both powder XRD and ¹H-¹³C FSLG HETCOR were able to detect changes in the sample.
- Phase transition of efivirenz from 1-EFV to β-EFV. Powder XRD and 19F MAS clearly confirmed the phase transition. Interestingly, this transition was not observed when 1-EFV did not contain any impurities of β-EFV before MAS or when 1-EFV was mixed with excipients in the marketed formulation of efivirenz.
Conclusions
Solid-state NMR is an excellent technique to study APIs and pharmaceutical products in solid state. It is a non-destructive technique. However, it has been shown that MAS can induce phase transitions and structural rearrangements in amorphous and crystalline APIs. Spinning sensitive samples at maximum spin rates allowed by your probe may not be the best way. Rather, spinning samples at rates that are really required and only for so long as required, may help. There are special pulse sequences for spinning sideband suppression and homonuclear decoupling sequences for resolution enhancement at slow and moderate spin rates. Finally, ¹H-¹³C FSLG HETCOR can detect changes in samples even if 1D NMR techniques and powder XRD fail.
If you wish to know more about JEOL ECZ Luminous NMR spectrometers and solid-state NMR probes, click the links. A brochure on JEOL solid-state NMR products and applications is available here.

{kind=link}
{kind=link}
{kind=link}